FAIR eDNA (FAIRe) guidelines
In this section, we outline the various data components and formats, present a comprehensive FAIR eDNA (FAIRe) metadata checklist (click here to download) along with scripts and tools to guide users through the data formatting process.
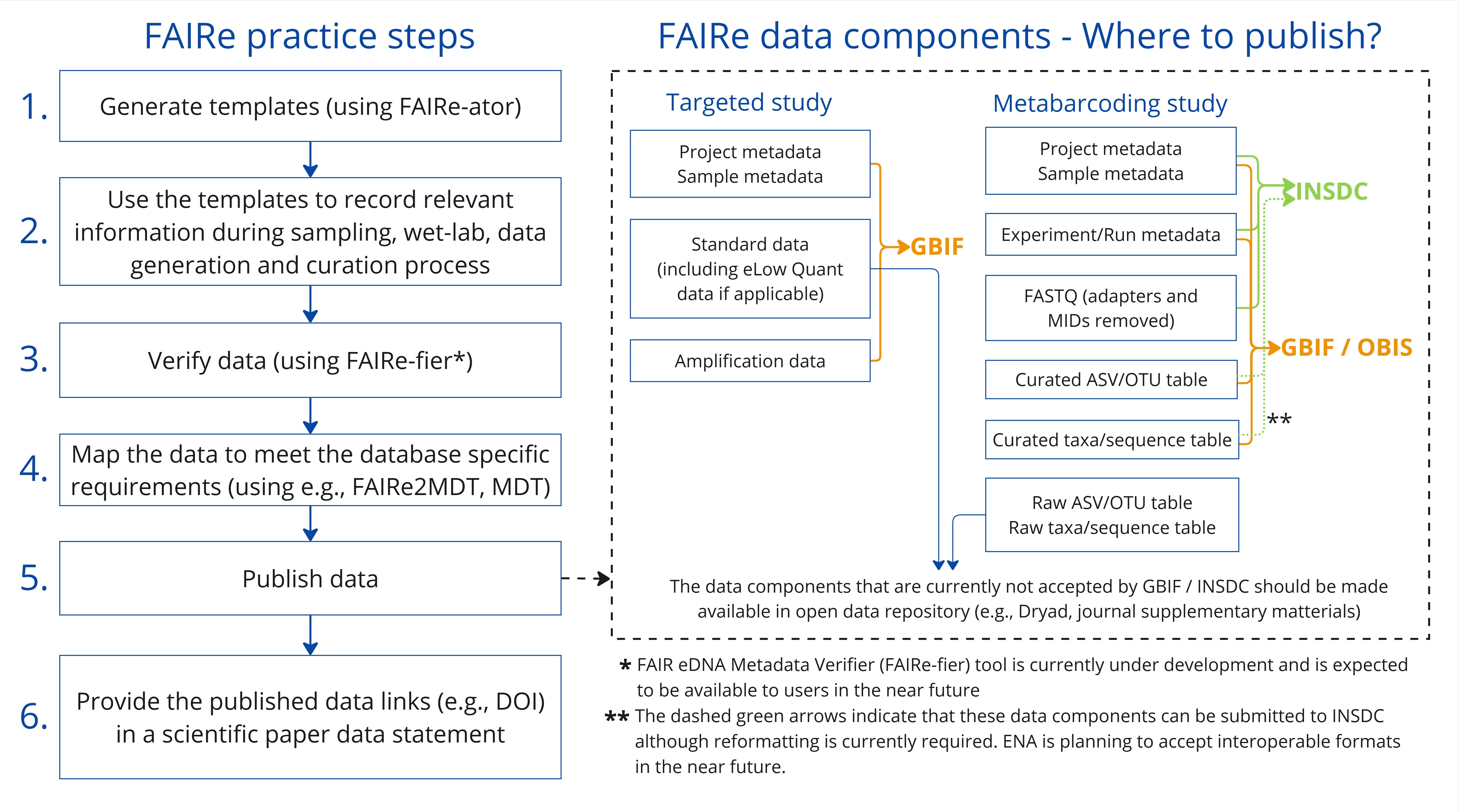
Data formats and components
The figure and list below outline the associated eDNA data components as well as their contents and purposes. In our proposed data structure, all data components, except DNA sequence data (i.e., raw data in FASTQ and, optionally, representative sequences in FASTA), are organised in a tabular format. They can be saved as individual worksheets in spreadsheet workbooks, as character-separated values text files (tab or comma), or as a combination of these (see various example datasets provided). This allows data to be conceptually divided into separate modules that can be linked using unique identifiers, such as project_id, sample_name, and assay_name, as well as to accommodate users’ needs and data wrangling preferences.
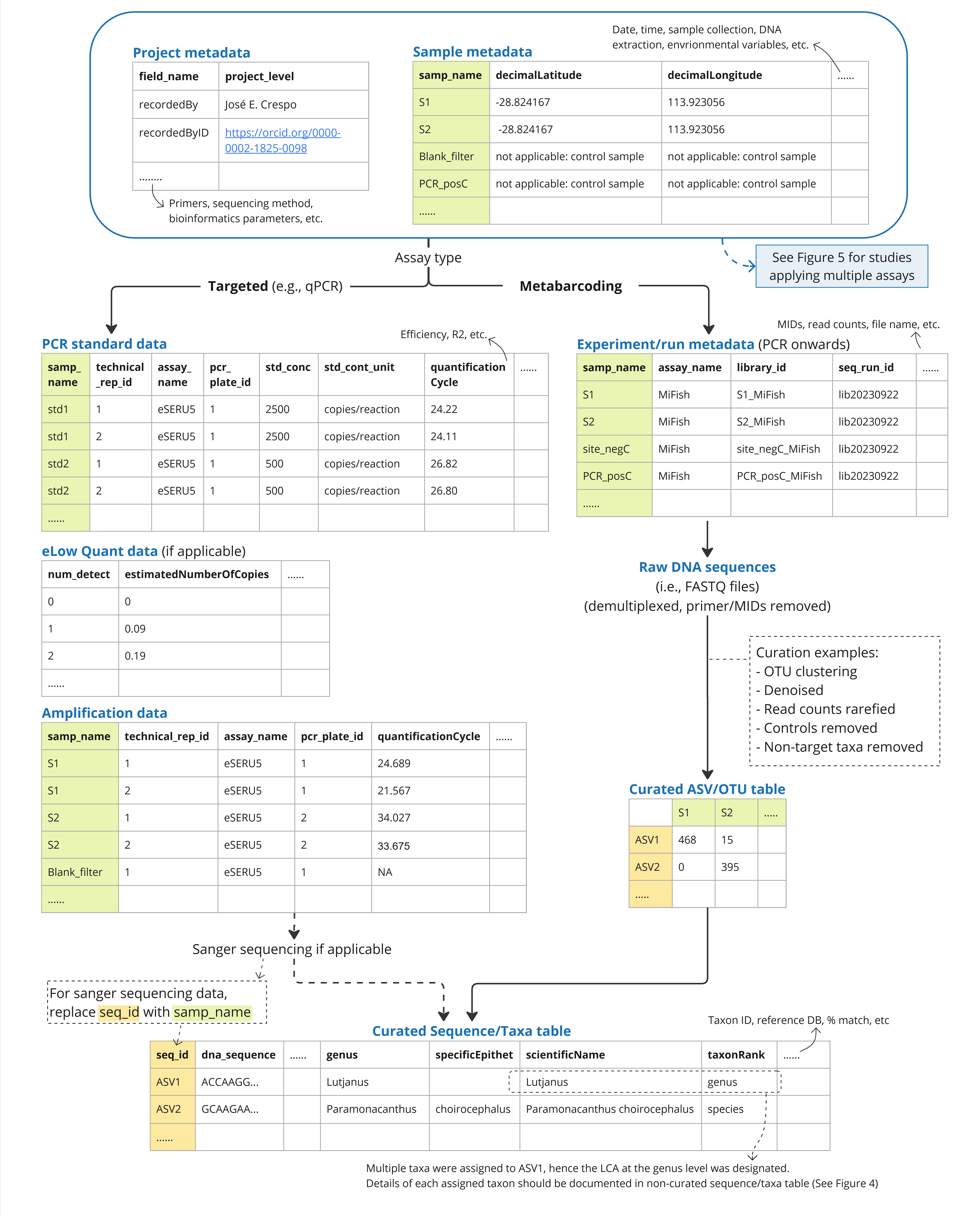
-
Project metadata (projectMetadata)
- Contents: Information that applies to an entire project and dataset. This includes a project name, reference associated to the datasets from which the datasets were derived (e.g., bibliographic references to studies, DOI of published, associated data), and metadata describing workflows including PCR, sequencing, and bioinformatic steps.
- Purpose: To describe project information and methodologies that apply to all samples, thereby avoiding the need to propagate identical entries to all samples in sample metadata. Information, such as bioinformatic quality filtering parameters and thresholds, enable data reusers to assess whether the data quality meets the specific requirements for certain types of reuse. Standardisation of such information is particularly important as minimum requirements for amplification analysis, bioinformatic workflows and quality assurance levels vary between studies and applications, including those reusing shared data.
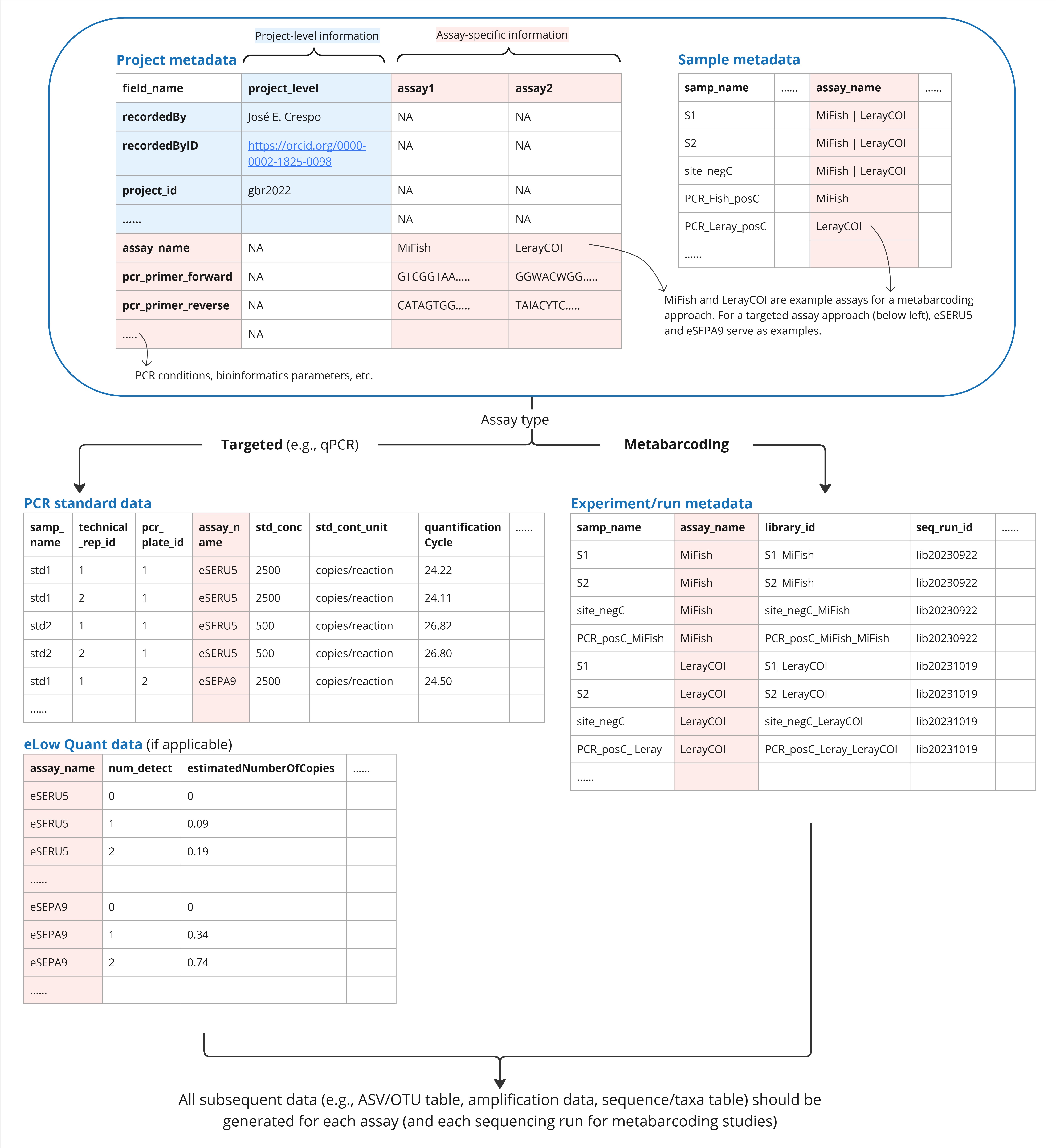
- Note: Multiple primer sets are often applied within one project to target more than one taxon. In this case, practitioners would follow the format in Figure 5 to record assay-specific workflows and outputs.
-
Sample metadata (sampleMetadata)
- Contents: Information for each sample (where this cannot be stated at the project level in project metadata), including sampling date, location, methodologies, and environmental variables (i.e., temperature, water depth).
- Purpose: To record sample-specific information through the workflows from sampling to bioinformatics.
-
Experiment/run metadata (experimentRunMetadata)
- Contents: Sample-specific information for PCR, library preparation and sequencing workflows.
- Purpose: To record workflows, from PCR onward, that are specific to each sample and assay.
- Note: Each entry (row) has a unique library ID (
lib_id) and multiplexing identifiers (MIDs) (mid_forward,mid_reverse).
-
PCR standard data (stdData)
- Contents: Detailed information of PCR standards, including starting input quantity, Ct/Cq values, and standard curve parameters (e.g., efficiency, R2).
- Purpose: To improve reproducibility and allow data reusers to regenerate the estimated copy numbers using their own approach. The entries in
assay_nameandpcr_plate_idin the PCR standard data and amplification data allow linking of the standards and eDNA samples.
-
eLow Quant data (eLowQuantData)
- Contents: Outputs from the eLow Quant binomial approach for PCR standards if applicable (see Lesperance et al., 2021).
- Purpose: To improve reproducibility and allow data reusers to regenerate the estimated copy numbers using their own approach.
-
Targeted assay amplification data (ampData)
- Contents: Raw amplification data of each PCR technical replicate performed on eDNA samples, as well as estimated copy numbers based on standard curves (if applicable) and detection status of targeted taxon in each biological replicate (field sample).
- Purpose: The Ct/Cq values of each technical replicate, combined with the standard information above, enable the re-conversion of the raw amplification data to concentration or detection status in future studies. Detect/non-detect records of a target taxon at a biological replicate level allow prompt reuses of taxon occurrence records in future studies, combined with sample metadata. They also standardise eDNA data in the context of biodiversity data produced using other methods, and consequently make the collective data and underlying research discoverable and reusable for wider audiences.
- Note: "NA" (not zero) should be used under the term
quantificationCycle(i.e., Ct/Cq values) to indicate no amplification occurred, because zero could be interpreted as an extremely high DNA concentration, which may or may not be target DNA, that would be unquantifiable due to being outside the standard curve range.
-
Raw DNA sequences
- Contents: DNA sequences in FASTQ format, demultiplexed for each sample. The sequences of primers, adapters and MIDs must be removed.
- Purpose: To allow data reusers to repeat quality filtering, denoising and taxonomic assignment using their own algorithms and thresholds to fit their purposes. To enable re-analyses using different or improved pipelines/algorithms. To enhance the reproducibility of studies.
-
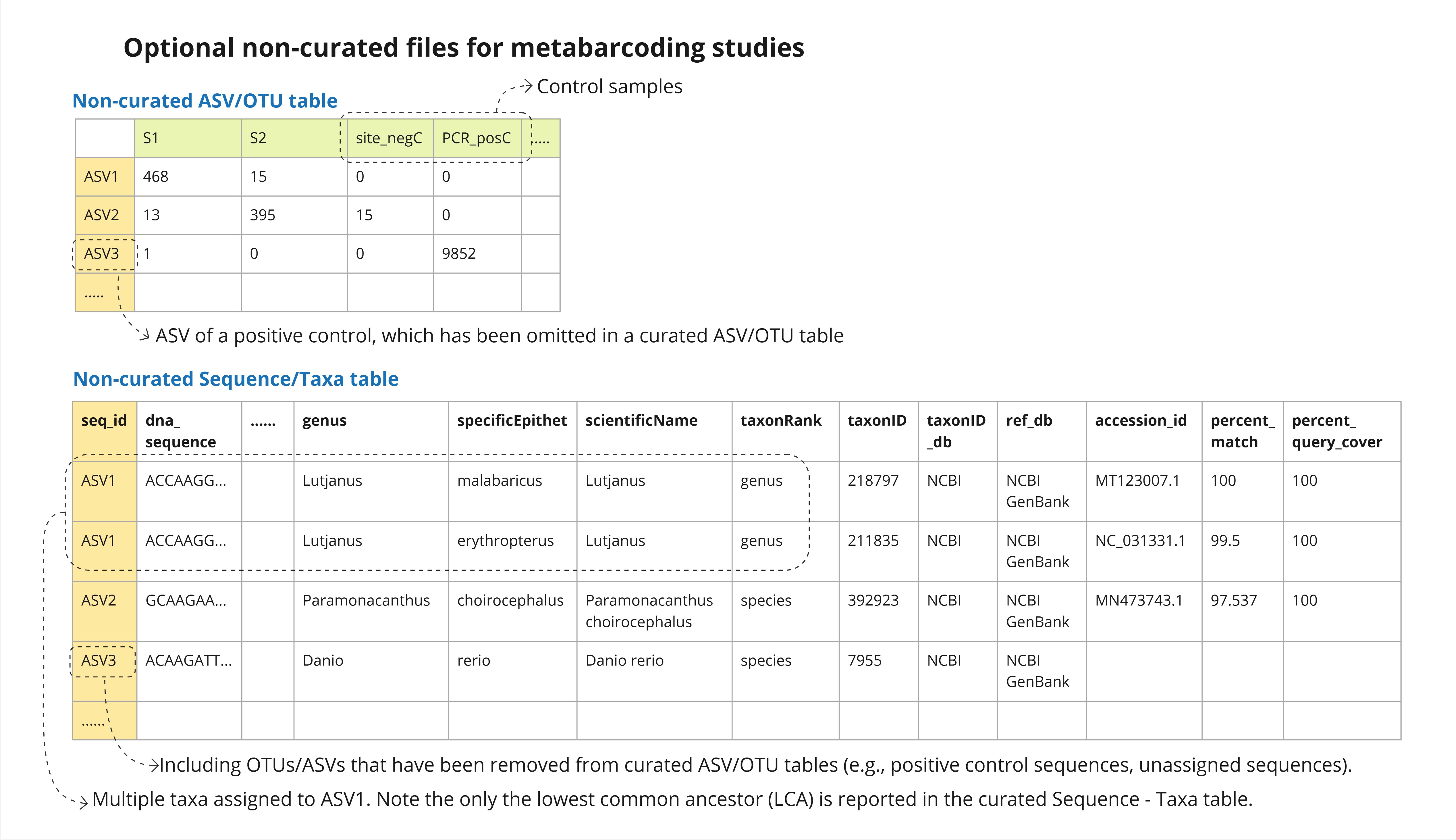
Non-curated ASV/OTU table (otuRaw) (Optional)
- Contents: The initial ASV/OTU table produced in a bioinformatic pipeline, containing absolute sequence read counts of all ASV/OTUs (including contaminant/non-targeted taxa and unassigned ASVs/OTUs) across all samples (including controls). Each ASV/OTU will be provided with a unique
seq_idat this stage. - Purpose: To communicate objective information of read counts of all OTUs/ASVs in all samples, and allow data reusers to modify or re-run the data curation steps if required. The inclusion of control samples, suspected contaminants and non-targeted taxa allows the investigations of common contamination sequences, taxa, and sources across multiple studies, and potentially identify how to minimize them.
- Note: The column names in ASV/OTU tables should be either
samp_nameorlib_id, making sure they maintain clear links between the experiment/run metadata and ASV/OTU table. - Note: Although it is called a “raw” ASV/OTU table, some level of compression, quality filtering and curation may already have been applied, such as filtering based on minimum length, read counts, error rates, and OTU clustering. The extent of data curation performed on the non-curated ASV/OTU table can vary between studies depending on the bioinformatics pipeline used, and this should be described under the term
otu_raw_descriptionin the project metadata.
- Contents: The initial ASV/OTU table produced in a bioinformatic pipeline, containing absolute sequence read counts of all ASV/OTUs (including contaminant/non-targeted taxa and unassigned ASVs/OTUs) across all samples (including controls). Each ASV/OTU will be provided with a unique
-
Curated ASV/OTU table (otuFinal)
- Contents: An ASV/OTU table resulting from various data curation processes, including denoising (i.e., custom or LULU curation), removing control samples, and eliminating suspected contamination and non-target taxa. This table should ideally be identical to, or correspond closely to, the one used for the final (e.g. ecological) analyses published in associated scientific papers. ASVs/OTUs without taxonomic assignments should remain in the otuFinal table if they were relevant to the analyses.
- Purpose: To facilitate reproducibility of original (ecological) analyses, and general reuse of a curated/cleaned version of the data. To improve interoperability with other types of biodiversity data, and thereby facilitate incorporation in general biodiversity databases, and make the data and underlying research discoverable and reusable for wider audiences.
- Note: The column names in ASV/OTU tables should be either
samp_nameorlib_id, making sure they maintain clear links between the experiment/run metadata and ASV/OTU table. - Note: In some studies, researchers may choose to collapse multiple ASVs/OTUs assigned to the same taxon, summing their read counts. We, however, strongly encourage data providers to submit ASV/OTU tables in their original, non-collapsed form. Preserving each ASV/OTU with its individual read counts and sequence ensures better interoperability and prevents the loss of potentially valuable ASVs/OTUs due to uncertain or erroneous taxonomic annotations resulting from incomplete or imperfect reference databases at the time of analysis.
- Note: Descriptions on how data were curated to produce the curated ASV/OTU table should be recorded under the term
otu_final_descriptionin the project metadata.
-
Non-curated Sequence/Taxa table (taxaRaw) (Optional)
- Contents: In addition to the contents in the curated Sequence - Taxa table (below), this table includes ASVs/OTUs that were excluded from the curated ASV/OTU table (e.g., non-target taxa, contaminants, positive control sequences, ASVs with no taxa hits). It may also contain multiple rows for a single ASV/OTU assigned to multiple taxa, with information of each assigned taxon.
- Purpose: To enhance data transparency by providing access to ASVs, OTUs, and taxa excluded from the curated ASV/OTU table. To allow data reusers the opportunity to re-assess and re-run specific data curation steps.
- Note: When assigning a lowest common ancestor (LCA) to a single ASV/OTU, multiple rows should be generated for the ASV/OTU to capture information about each of the assigned taxa. Users may apply a similarity threshold (e.g., 97%) and include all reference sequences within this range, or a representative subsample of them, in the output.
- Note: See the example in Figure 3 (ASV1 in the non-curated Sequence/Taxa table).
-
Curated Sequence/Taxa table (taxaFinal)
- Contents: DNA sequence of each
seq_idlisted in the curated ASV/OTU table. If a taxon was assigned, also include assigned taxonomy and assignment quality assurance parameters (e.g., % identity, % query coverage). If multiple taxa are assigned to a single ASV/OTU, the LCA should be used as the assigned taxon. - Purpose: To allow data reusers to assess the specificity and accuracy of the inferred taxonomy, and to perform taxonomic re-annotation using different or updated sequence reference databases.
- Note: ASVs/OTUs without taxonomic assignments should be included in the taxaFinal table, but without taxonomic information (e.g.,
scientificName), if they were relevant to the study scope.
- Contents: DNA sequence of each
Consistent identifiers and file naming
To ensure machine readability and long-lasting reference to a digital resource, it is crucial to apply consistent, persistent sample and sequence identifiers (samp_name, lib_id and seq_run_id) across datasets (Damerow et al., 2021; McMurry et al., 2017). Similarly, each file must have a clear, unambiguous name, consisting of project_id, assay_name and seq_run_id (see Table 2 and example datasets) to maintain clear links between files. For example, a curated ASV/OTU table should be named as otuFinal_project_id_assay_name_seq_run_id (e.g., otuFinal_gbr2022_MiFish_lib20230922.csv). If multiple data components are stored within a single spreadsheet workbook, the file name should be the project_id (e.g., gbr2022.xlsx). In this case, each worksheet name should follow the format in Table 2 without the project_id as it already appears in the main file name (e.g., otuFinal_MiFish_lib20230922).
Table 1. Standardised names for each data type. In the examples, project_id = gbr2022, samp_name = S1, assay_name = eSERUS and MiFish for targeted and metabarcoding assay approaches respectively, seq_run_id = run20230922
| Data type | Name format | Example |
|---|---|---|
| Project metadata | projectMetadata_project_id |
projectMetadata_gbr2022.csv |
| Sample metadata | sampleMetadata_project_id |
sampleMetadata_gbr2022.csv |
| PCR standard data | stdData_project_id |
stdData_gbr2022.csv |
| eLow Quant data | eLowQuantData_project_id |
eLowQuantData_gbr2022.csv |
| Amplification data | ampData_project_id_assay_name |
ampData_gbr2022_eSERUS.csv |
| Experiment/run metadata | experimentRunMetadata_project_id |
experimentRunMetadata_gbr2022.csv |
| Non-curated ASV/OTU table | otuRaw_project_id_assay_name_seq_run_id |
otuRaw_gbr2022_MiFish_run20230922.csv |
| Curated ASV/OTU table | otuFinal_project_id_assay_name_seq_run_id |
otuFinal_gbr2022_MiFish_run20230922.csv |
| Non-curated Sequence/Taxa table | taxaRaw_project_id_assay_name_seq_run_id |
taxaRaw_gbr2022_MiFish_run20230922.csv |
| Curated Sequence/Taxa table | taxaFinal_project_id_assay_name_seq_run_id |
taxaFinal_gbr2022_MiFish_run20230922.csv |
FAIRe metadata checklist
We developed a FAIR eDNA (FAIRe) metadata checklist, providing a comprehensive vocabulary for describing different data components and methodologies. Visit the Download tab to access the latest version of the checklist, the full template, and the previous versions with change logs. Thorough documentation is crucial for improving the transparency and reproducibility of studies, as well as for enabling the evaluation of data suitability for specific reuse cases. The FAIRe checklist consists of 337 data terms (38 mandatory, 51 highly recommended, 128 recommended and 120 optional terms), organised into workflow sections (e.g., sample collection, PCR, bioinformatics). Most of the mandatory, highly recommended and recommended information is generated naturally during the course of any project. As a result, data meeting the minimum reporting requirements can be readily made available and submitted to public repositories.
Vocabulary basis
The FAIRe metadata checklist consists of terms sourced from existing data standards such as MIxS, DwC, and the DNA-derived data extension to DwC. MIxS offers several checklists and extensions relevant for eDNA data, including Minimum Information about a Marker Sequence (MIMARKS), Minimum Information about Metagenome or Environmental Sequence (MIMS), and extensions for various environments (e.g., water, sediment, soil, air, host associated, and symbiont associated). These standards were reviewed and incorporated into the FAIRe metadata checklist where applicable to ensure comprehensive coverage of eDNA data needs. We have also incorporated terms from MIQE guidelines (Bustin et al., 2009), the single species eDNA assay development and validation checklist (Thalinger et al., 2021) and Minimum Information for eDNA and eRNA Metabarcoding (MIEM) guidelines (Klymus et al., 2024). The source and URI for each term from existing standards are documented in the FAIRe metadata checklist under the columns ‘source’ and ‘URI’. Where necessary, modifications have been made to term names, descriptions, and examples for easier interpretation by the eDNA community. A total of 158 new terms were proposed to accommodate some diverse attributes of eDNA procedures and datasets, and to improve the possibility of evaluating fitness for reuse. Among these are terms related to targeted assay detection workflows (e.g., lod_method, std_seq), bioinformatic tools, filtering parameters and cutoffs (e.g., demux_tool, demux_max_mismatch), taxonomic assignment metrics (e.g., percent_match), and taxon screening methods (i.e., screen_contam_method, screen_nontarget_method). The FAIRe checklist also includes a range of environmental variables relevant to eDNA samples, sourced from the MIxS sample extensions including Water, Soil, Sediment, Air, HostAssociated, MicrobialMatBiofilm and SymbiontAssociated. These variables are typically recorded during sampling events, and sharing them enables more advanced modelling approaches among other applications.
If suitable term names are not available in the FAIRe checklist , users should search for them in existing standards, such as MIxS and DwC, and use these standardised terms where possible. If relevant terms cannot be found in these resources, users may add new terms using clear, concise, and descriptive names within related tables.
Specified use of terms and controlled vocabularies
Several approaches were implemented to increase standardisation of terms. Firstly, for some free-text terms from existing standards, we propose using controlled vocabularies. For example, the value for the target_gene term should be selected from a list of 28 gene regions used for barcoding. This standardisation prevents variations of COI such as CO1, COXI, COX1, Cytochrome oxidase I gene, Cytochrome c oxidase I, and spelling mistakes/variants of these from being entered. If a suitable value cannot be found among the specified values of the vocabulary, it is possible to enter “other:” followed by a free-text description. Secondly, units of numeric variables are strictly specified based on the International System of Units wherever possible, and only numeric entries without units are allowed. When units are necessary for unambiguous communication, separate terms for measurement value and measurement unit are provided, with the unit restricted to a controlled vocabulary (e.g., samp_size for the numeric value and samp_size_unit with options of mL, L, mg, g, kg, cm2, m2, cm3, m3, and other). Thirdly, formats for some data terms are restricted, generally following other standards (i.e., DwC, MIxS) and data infrastructures (i.e., GBIF, OBIS, INSDC). For example, decimalLatitude and decimalLongitude are required terms, and must be in decimal degrees using WGS84 datum. If records in an original datasheet follow other formats (e.g., degree minute second, UTM, datums other than WGS84), they should be stored under the terms verbatimLatitude and verbatimLongitude while decimalLatitude and decimalLongitude are also required to be entered. Similarly, sampling date and time are summarized under the term eventDate,
which utilises the ISO 8601 format, followed by the difference from UTC time (e.g., "2008-01-23T19:23-06:00" in the time zone six hours earlier than UTC). Original records of date and time before conversion into the ISO 8601 format should be stored under the terms verbatimDate and verbatimTime.
Missing values
Information may be missing for various reasons, and historically, science has used a variety of conventions to denote missing data. Furthermore, geographical coordinates can be generalised or withheld to protect endangered species or culturally important sites for Indigenous People and Local Communities (Chapman, 2020; Chapman and Wieczorek, 2020; Frank et al., 2015, https://fnigc.ca/). The reasons for withholding or generalising data should be described under the terms informationWithheld and dataGeneralization respectively in project metadata. When a value is missing from a mandatory term, it is required to provide the reason following the format of the INSDC missing value controlled vocabulary. It is also recommended to apply this approach for non-mandatory terms.
Below, we provide the complete list of missing value terms, including their full hierarchical structure for clarity. Only the bold texts should be used to describe the missing values (i.e., do not include the parenthetical explanations or add your own reasons).
- not applicable: control sample
- not applicable: sample group
- not applicable (other reasons not applicable)
- missing: not collected: synthetic construct
- missing: not collected: lab stock
- missing: not collected: third party data
- missing: not collected (other reasons not collected)
- missing: not provided: data agreement established pre-2023
- missing: not provided (other reasons not provided)
- missing: restricted access: endangered species
- missing: restricted access: human-identifiable
- missing: restricted access (other reasons restricted access)
Data submission/publication
Publishing eDNA datasets to databases such as GBIF, OBIS and INSDC, is an essential step in ensuring FAIR data practices. These platforms ensure data findability and accessibility for scientific research and decision-making that rely on open data, either through application programming interfaces (APIs) or sophisticated web-browser interfaces. They offer data validation procedures during submission, and provide additional standardisation to ensure interoperability. Persistent sample and sequence identifiers are provided upon the submission of data to these databases, which are essential for machine-readability and long-lasting reference to the data. For example, in GBIF, dataset authors and publishers receive credit through unique dataset Digital Object Identifiers (DOIs) that enable citation tracking and automatic usage reporting. Since not all eDNA data components can be sufficiently accommodated in any single of these databases, they should be submitted in parts to the most suitable databases (Figure 2). These are the current recommended practices for sharing FAIR eDNA data through the larger databases/infrastructures:
- Submit raw DNA sequence data and metadata to nucleotide databases such as INSDC (ENA, NCBI or DDBJ). For instance, NCBI allows submission of project metadata via BioProject, sample metadata via BioSample, and raw sequences via the Sequence Read Archive (SRA) (Barrett et al., 2012). This links metadata with sequence data, enabling users to retrieve complete datasets by querying BioProject or BioSample records.
- Publish the derived biodiversity data – i.e., inferred taxon occurrences with sequences and metadata – to biodiversity databases such as GBIF, or GBIF and OBIS (if marine data). Both GBIF and OBIS rely on data being formatted according to the DwC Standard with the DNA-derived data extension as described in the co-authored guide (Abarenkov et al., 2023). The dataset is converted into so-called Darwin Core Archive (DwC-A) that can be indexed by GBIF and OBIS.
- Archive the remaining data components (i.e., standard data for targeted assay studies and raw ASV/OTU table for metabarcoding studies), formatted according to the FAIRe guidelines, in open data repositories such as Dryad, Zenodo and Figshare or as supplementary materials in journals. This approach ensures comprehensive storage of raw data which are not addressed in the above steps. It is, however, essential to recognise that datasets in these repositories are not indexed to the extent required for effective searchability and interoperability within larger eDNA, biodiversity, or nucleotide data platforms.
- Publishing protocols and sharing bioinformatics and analytical codes is also highly recommended (Jenkins et al., 2023; Teytelman et al., 2016) through open-source platforms such as protocols.io and WorkflowHub.
- All above data, protocols and codes should be assigned DOIs, which should be documented in data accessibility statements within journal articles, as well as in the metadata terms such as
seq_archive,code_repoandassociated_resource.
The guidelines provided here result in data formatted slightly different from those accepted by nucleotide and biodiversity databases. Hence, minor reformatting is required. One example is the necessary data format when multiple assays were applied in a single project. GBIF recently launched the Metabarcoding Data Toolkit (MDT), a user-friendly web application that reshapes tabular metabarcoding data—similar to the format proposed here—and publishes it to GBIF and OBIS complying with the guidelines in Abarenkov et al. (2023). The GBIF MDT currently requires a separate input dataset for each assay (hence information from project and sample metadata are repeated), whereas the FAIRe checklist allow the metadata from multiple assays to be combined (Figure 5). An R script (FAIRe2MDT) has been developed to convert FAIRe data templates for GBIF and OBIS publication via MDT, bridging the differences in data formats between FAIRe and GBIF standards (see below section ‘Available scripts and tools’). Using these tools in combination streamlines the publication of eDNA data to GBIF and OBIS. Overall, there is currently a strong effort by biodiversity data platforms to improve the suitability of depositing eDNA-based data.
Available scripts and tools
The below scripts and tools were developed to aid data formatting according to the guidelines, and are made available at the FAIRe github repositories.
- FAIRe-ator (FAIR eDNA template generator): This R function creates data templates based on user-specified parameters, such as assay type (i.e., targeted or metabarcoding assay), sample type (e.g., water, sediment), and the number of assays applied. Additionally, the function allows users to input project ID and assay name(s), which ensures correct file name formatting and pre-fills the
project_idandassay_nameterms in the template. Download the README and R code here. - FAIRe-fier (FAIR eDNA metadata verifier): This tool is accessible via web interface. Source code is on GitHub, and additional metadata and citation details can be found at http://hdl.handle.net/102.100.100/706519?index=1. It allows users to validate their metadata without requiring any scripting knowledge. After filling their project and sample metadata, users can upload it to the tool for validation. The tool checks various components, including whether mandatory terms are completed or, if not, whether a valid reason has been provided under the
information_withheldterm in the project metadata. It also verifies that controlled vocabulary entries and fixed-format terms, such aseventDate(which must follow ISO 8601 format), are correctly formatted. If any issues are found, users receive output with warning and error messages, indicating where corrections are needed. Formatted output will be produced when there is no warning message. - FAIRe2MDT: The R script to convert FAIR eDNA data templates for GBIF submission via MDT. Download the R code here.
Tools Developed by the Community We’re excited to showcase a growing set of scripts and tools developed by FAIRe implementors! It’s fantastic to see more groups adopting the FAIRe guidelines and building automation tools to streamline their workflows. We are currently developing these tools in both R and Python as part of a new package – stay tuned via FAIRe Coding on the News & Updates page!
*If your community has developed FAIRe-related tools, please contact us – we’d love to connect and explore collaboration opportunities!
- FAIReSheets (FAIR eDNA template generator for Google Sheets): This python script creates the FAIRe eDNA Data Template in Google Sheets. It replicates the template creation from the FAIRe-ator, except FAIReSheets outputs the template to Google Sheets rather than a Microsoft Excel spreadsheet. Developed by Bayden Willms (ORCID/ GitHub) at NOAA AOML 'Omics
- FAIRe2ENA : Convert FAIRe-formatted metadata to ENA (European Nucleotide Archive) XML format for submission. Developed by Philipp Bayer (ORCID/ GitHub) at Minderoo Foundation OceanOmics
- Amplicon Nextflow : A bioinformatics pipeline for quality controls, denoising, ASVs and ZOTUs, taxonomic assignment, with an option to use FAIRe metadata as an input and output. Developed by Adam Bennett (ORCID/ GitHub), Philipp Bayer (ORCID/ GitHub), Sebastian Rauschert (ORCID/ GitHub) at Minderoo Foundation OceanOmics
Example datasets
Various example datasets are available to provide eDNA practitioners with suggestions for how to format and publish their data.
Below is a list of example datasets using targeted assays, formatted according to the FAIRe checklist v1.0. These datasets are available here.
| recordedBy | Mark Louie Lopez | Neha Acharya-Patel | Cecilia Villacorta-Rath | Lynsey Harper |
|---|---|---|---|---|
| project_id | AEP_Fish_sedDNA | Rockfish_targeted_qPCR | EirwiniBurdekin | NECR534_GCN_single-species |
| bibliographicCitation | DOI | DOI | DOI | Link |
| assay_type | targeted | targeted | targeted | targeted |
| assay_name | eFish1 | eESLU1 | eCOAR7 | eSEMA3 | eSEPA9 | eSERU5 | EirwiniND4 | TCCB |
| env_medium | lake sediment [ENVO:00000546] | sea water [ENVO:00002149] | river water [ENVO:01000599] | liquid water [ENVO:00002006] |
| Output file 1 | AEP_Fish_sedDNA.xlsx | Rockfish_targeted_qPCR.xlsx | EirwiniBurdekin.xlsx | NECR534_GCN_single-species.XLSX |
| Excel worksheet 1 | projectMetadata | projectMetadata | projectMetadata | projectMetadata |
| Excel worksheet 2 | sampleMetadata | sampleMetadata | sampleMetadata | sampleMetadata |
| Excel worksheet 3 | stdData | stdData | ampData | stdData |
| Excel worksheet 4 | eLowQuantData | eLowQuantData | ampData | |
| Excel worksheet 5 | ampData_eFish1 | ampData_eSERU5 | ||
| Excel worksheet 6 | ampData_eESLU1 | ampData_eSEPA9 | ||
| Excel worksheet 7 | ampData_eCOAR7 | ampData_eSEMA3 |
A list of example datasets using metabarcoding assays, formatted according to the FAIRe checklist v1.0. These datasets are available here.
| recordedBy | Bruce Deagle | Luke Thompson | Nick Dunn | Katrina West | Rachel Haderlé |
|---|---|---|---|---|---|
| project_id | CPR_2015 | noaa-aoml-gomecc4 | NECR534_GCN_metabarcoding | IOT-eDNA | verte_quadeloupe |
| bibliographicCitation | DOI | DOI | Link | West et al., in prep | DOI |
| assay_type | metabarcoding | metabarcoding | metabarcoding | metabarcoding | metabarcoding |
| assay_name | LerayCOI | ssu16sv4v5-emp | ssu18sv9-emp | 12S-V5 | 16SFish | COILeray | vert01 | tele01 | mamm01 | cetac |
| env_medium | planktonic material [ENVO:01000063] | sea water [ENVO:00002149] | marine sediment [ENVO:03000033] | liquid water [ENVO:00002006] | sea water [ENVO:00002149] | water surface [ENVO:01001191] |
| Output file 1 | CPR_2015.xlsx | noaa-aoml-gomecc4.xlsx | NECR534_GCN_metabarcoding.xlsx | IOT-eDNA.xlsx | verte_quadeloupe.xlsx |
| Output file 2 | outRaw_* (for each assay and sequencing run) | otuFinal_* (for each assay and sequencing run) | |||
| Output file 3 | outFinal_* (for each assay and sequencing run) | taxaFinal_* (for each assay and sequencing run) | |||
| Output file 4 | taxaRaw_* (for each assay and sequencing run) | ||||
| Output file 5 | taxaFinal_* (for each assay and sequencing run) | ||||
| Excel worksheet 1 | projectMetadata | projectMetadata | projectMetadata | projectMetadata | projectMetadata |
| Excel worksheet 2 | sampleMetadata | sampleMetadata | sampleMetadata | sampleMetadata | sampleMetadata |
| Excel worksheet 3 | experimentRunMetadata | experimentRunMetadata | experimentRunMetadata | experimentRunMetadata | otuFinal_* (for each assay and sequencing run) |
| Excel worksheet 4 | otuFinal | otuFinal | taxaFinal_* (for each assay and sequencing run) | ||
| Excel worksheet 5 | taxaFinal | taxaFinal |
Frequently Asked Questions
Q. What should I do with ASVs that have no BLAST hits?
A. ASVs without taxonomic hits are still valuable (e.g., for future re-annotation as reference databases improve). They should be included in the otuRaw and taxaRaw tables without taxonomic fields such as scientificName. If these ASVs fall within the scope of your study and are relevant to ecological analyses, they should also be retained in otuFinal and taxaFinal.
Q. Can FAIRe-fier verify all data components? A. No. Currently, FAIRe-fier validates projectMetadata and sampleMetadata for both metabarcoding and targeted assay studies, but does not yet validate other components (e.g., experimentRunMetadata, ampData). Support for these additional components is planned for a future version of FAIRe-fier.
Useful resources
The current FAIRe frameworks were developed using the following resources as foundational starting points.
- Darwin Core Quick Reference Guide
- Minimum Information about any (x) Sequence (MIxS) standard
- Darwin Core extension of DNA derived data
- Abarenkov et al., (2023) Publishing DNA-derived data through biodiversity data platforms, v1.3. Copenhagen: GBIF Secretariat.
- The OBIS manual
- GBIF Metabarcoding Data Toolkit